
Disorders of the calcium-sensing receptor and partner proteins: insights into the molecular basis of calcium homeostasis
- 1Academic Endocrine Unit, Radcliffe Department of Medicine, University of Oxford, Oxford, UK
- 2Department of Musculoskeletal Biology, Institute of Ageing and Chronic Disease, University of Liverpool, Liverpool, UK
- Correspondence should be addressed to R V Thakker; Email: rajesh.thakker{at}ndm.ox.ac.uk
Abstract
The extracellular calcium (Ca2+o)-sensing receptor (CaSR) is a family C G protein-coupled receptor, which detects alterations in Ca2+o concentrations and modulates parathyroid hormone secretion and urinary calcium excretion. The central role of the CaSR in Ca2+o homeostasis has been highlighted by the identification of mutations affecting the CASR gene on chromosome 3q21.1. Loss-of-function CASR mutations cause familial hypocalciuric hypercalcaemia (FHH), whereas gain-of-function mutations lead to autosomal dominant hypocalcaemia (ADH). However, CASR mutations are only detected in ≤70% of FHH and ADH cases, referred to as FHH type 1 and ADH type 1, respectively, and studies in other FHH and ADH kindreds have revealed these disorders to be genetically heterogeneous. Thus, loss- and gain-of-function mutations of the GNA11 gene on chromosome 19p13.3, which encodes the G-protein α-11 (Gα11) subunit, lead to FHH type 2 and ADH type 2, respectively; whilst loss-of-function mutations of AP2S1 on chromosome 19q13.3, which encodes the adaptor-related protein complex 2 sigma (AP2σ) subunit, cause FHH type 3. These studies have demonstrated Gα11 to be a key mediator of downstream CaSR signal transduction, and also revealed a role for AP2σ, which is involved in clathrin-mediated endocytosis, in CaSR signalling and trafficking. Moreover, FHH type 3 has been demonstrated to represent a more severe FHH variant that may lead to symptomatic hypercalcaemia, low bone mineral density and cognitive dysfunction. In addition, calcimimetic and calcilytic drugs, which are positive and negative CaSR allosteric modulators, respectively, have been shown to be of potential benefit for these FHH and ADH disorders.
Introduction
Extracellular calcium (Ca2+o) is essential for the regulation of a variety of biological processes including neuromuscular excitability, blood coagulation and mineralisation of bone matrix and for providing a steady supply of calcium for intracellular functions ranging from muscle contraction to hormone synthesis and secretion (Brown 1991). Thus, there is a requirement for Ca2+o to be tightly regulated, and a complex homeostatic system has evolved to maintain Ca2+o at near-constant concentrations (Fig. 1) (Thakker 2016). The Ca2+o homeostatic system comprises four main components: (1) the parathyroid glands, which sense the prevailing Ca2+o concentrations and regulate the actions of target calcitropic organs; (2) the intestine and kidneys, which facilitate the transfer of calcium between the external environment and extracellular fluid; (3) the skeleton, which represents the major reservoir of total body calcium and is critical for buffering short-term changes in Ca2+o concentrations; and (4) calcitropic hormones such as parathyroid hormone (PTH) and 1,25-dihydroxyvitamin D3, which mediate the interactions between the parathyroid glands, bone, kidney and intestines (Fig. 1). Thus, low Ca2+o concentrations lead to PTH release from the parathyroid glands. This hormone exerts three distinct effects on Ca2+o homeostasis, which are to enhance bone resorption, urinary calcium reabsorption, and the renal synthesis of 1,25-dihydroxyvitamin D3, thereby leading to intestinal calcium absorption (Fig. 1). Low Ca2+o concentrations are also sensed by the kidneys, which increase urinary calcium reabsorption independent of the actions of PTH (Loupy et al. 2012, Riccardi & Valenti 2016). These combined events mediate a rise in Ca2+o concentrations, which together with 1,25-dihydroxyvitamin D3, leads to feedback inhibition of PTH secretion (Fig. 1) (Thakker 2016).
Overview of Ca2+o homeostasis. The parathyroid CaSR senses reductions in Ca2+o, which leads to a rapid rise in PTH secretion. The increased circulating PTH acts via the PTH1-receptor (PTH1R) in the kidneys and bone. The skeletal effects of PTH are to increase bone resorption, thereby releasing calcium into the extracellular fluid. In the kidney, PTH increases calcium reabsorption and stimulates the proximal renal tubular 1-α-hydroxylase (1αOHase) enzyme, which promotes the synthesis of the active 1,25-dihydroxyvitamin D3 (1,25D3) metabolite from 25-hydroxyvitamin D3 (25D3), which is the major circulating form of vitamin D. The elevated 1,25D3 acts on the intestine via the vitamin D receptor (VDR) to increase the absorption of dietary calcium. Thus, in response to hypocalcaemia, the secretion of PTH, by these direct and indirect actions leads to the restoration of normocalcaemia. The kidney CaSR senses reductions in Ca2+o and promotes urinary calcium reabsorption independent of the actions of PTH. The rise in Ca2+o and 1,25D3 concentrations mediated by PTH act on the parathyroid glands to induce feedback inhibition of further PTH secretion.
Studies of patients and kindreds with familial hypocalciuric hypercalcaemia (FHH) or autosomal dominant hypocalcaemia (ADH) have helped to elucidate the molecular basis of Ca2+o sensing and overall regulation of Ca2+o homeostasis by calcitropic tissues such as the parathyroid glands and kidneys. These studies also revealed the involvement of a G protein-coupled receptor (GPCR) pathway that comprises the calcium-sensing receptor (CaSR), G-protein α-11 (Gα11) subunit and adaptor-related protein complex 2 sigma (AP2σ) subunit (Pollak et al. 1993, Nesbit et al. 2013a,b). This article will provide an overview of the role of the CaSR, Gα11 and AP2σ proteins, review the disorders caused by mutations of these proteins, outline potential targeted therapies, and describe insights gained into the molecular basis of Ca2+o homeostasis by studies of the CaSR and its partner proteins.
Overview of calcium-sensing receptor signalling and trafficking
The human CaSR is a 1078 amino acid dimeric cell-surface protein that belongs to class C of the GPCR superfamily, which also includes the metabotropic glutamate, gamma-aminobutyric acid type B (GABAB) and taste 1 receptors (Katritch et al. 2013). The CaSR is highly expressed in the parathyroid glands and kidneys (Regard et al. 2008). It has a large (612 amino acid) extracellular domain (ECD), which was recently crystallised and shown to comprise two globular lobes that adopt a venus flytrap (VFT) conformation (Zhang et al. 2016). The cleft region located between the two lobes of the VFT is predicted to bind Ca2+o (Fig. 2) (Huang et al. 2007, Hannan et al. 2012), thereby leading to the activation of multiple intracellular signalling cascades via interactions with its transmembrane and intracellular domains (Conigrave & Ward 2013, Hofer & Brown 2003). In the parathyroid glands, the Gq/11 family of heterotrimeric guanine nucleotide–binding proteins (G proteins) are considered to represent the major downstream signalling partners for the CaSR (Varrault et al. 1995, Wettschureck et al. 2007, Nesbit et al. 2013a). The binding of the CaSR to the Gq and G11 proteins is predicted to lead to the dissociation of the respective Gα-subunits from their obligate Gβγ heterodimer, which activate the phospholipase C-beta (PLCβ) enzyme, thereby facilitating the hydrolysis of a plasma membrane lipid constituent, known as phosphatidylinositol 4,5-bisphosphate (PIP2), to produce inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG) (Fig. 2) (Hofer & Brown 2003, Conigrave & Ward 2013). IP3 in turn stimulates the rapid release of calcium from intracellular stores (Breitwieser & Gama 2001), whereas DAG activates the mitogen-activated protein kinase (MAPK) cascade (Corbetta et al. 2002). These intracellular events mediate a decrease in PTH secretion from the parathyroid chief cell and a reduction in renal tubular calcium reabsorption (Fig. 2). The level of CaSR cell-surface expression likely influences Ca2+o homeostasis and is regulated by agonist-driven insertional signalling (ADIS), which leads to enhanced anterograde trafficking of newly synthesised receptors to the plasma membrane after prolonged exposure to Ca2+o (Grant et al. 2011). Moreover, the β-arrestin protein together with the adaptor-related protein complex 2 (AP2), which comprises a heterotetrameric complex of α-, β-, μ- and σ-subunits (Nesbit et al. 2013b) is considered to facilitate CaSR internalisation from the plasma membrane by clathrin-mediated endocytosis and retrograde trafficking (Breitwieser 2013). Thus, AP2 and β-arrestin likely represent key determinants of CaSR cell-surface expression.
Role of the CaSR, Gα11 and AP2 complex in the regulation of PTH secretion and renal tubular calcium reabsorption. The binding of calcium (red filled-in circle, Ca) to the extracellular bilobed venus fly trap (VFT) domain of the CaSR (grey) results in Gα11 (yellow)-dependent stimulation of phospholipase C-β (PLCβ) (dark blue) activity, which catalyses the formation of inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG) from phosphatidylinositol 4,5-bisphosphate (PIP2). An accumulation of IP3 mediates the rapid release of calcium into the cytosol from intracellular stores, whereas DAG activates the MAPK cascade. These intracellular signalling events lead to a decrease in PTH secretion from the parathyroid chief cell and reduction in renal tubular calcium reabsorption. CaSR cellsurface expression is regulated by agonist-driven insertional signalling (ADIS) (not shown) (Grant et al. 2011) and also by an endocytic complex comprising clathrin, β-arrestin (green) and the AP2 complex (orange), which traffic this GPCR to the endosomal-lysosomal degradation pathway (light blue) or recycle the CaSR back to the cell surface (Breitwieser 2013). Loss- and gain-of-function mutations of the CaSR lead to FHH1 and ADH1, respectively, whereas loss- and gain-of function mutations of the Gα11-subunit are associated with FHH2 and ADH2, respectively. Loss-offunction mutations of the AP2σ-subunit lead to FHH3.
Disorders associated with calcium-sensing receptor mutations
More than 230 different germline mutations of the CaSR, which is encoded by the CASR gene (Fig. 3) located on chromosome 3q21.1, have been reported (Hannan & Thakker 2013). These mutations may cause a loss of CaSR function and give rise to hypercalcaemic disorders such as FHH type 1 (FHH1), neonatal severe hyperparathyroidism (NSHPT) and adult-onset primary hyperparathyroidism (PHPT); or lead to a gain of function that is associated with hypocalcaemic disorders such as ADH type 1 (ADH1) and Bartter syndrome type V (Table 1) (Hannan & Thakker 2013).
(A) Schematic representation of the genomic organisation of the CASR gene showing mutational hotspots for disease-associated missense mutations. The CASR gene consists of six coding exons (2–7), and the start (ATG) and stop (TGA) codons are in exons 2 and 7, respectively. The 5′ portion of exon 2 and the 3′ portion of exon 7 are untranslated (open boxes). The 3′ portion of exon 2, exons 3, 4, 5 and 6 and the 5′ portion of exon 7, encode the extracellular domain (light grey). The mid portion of exon 7 encodes the transmembrane (dark grey) and intracellular (black) domains. More than 170 different missense CASR mutations have been reported in patients with FHH1, NSHPT, adult-onset PHPT, ADH1 and Bartter syndrome type V (Hannan & Thakker 2013). These mutations affect >110 different codons scattered throughout the CASR gene. Twenty-eight codons (>20% of all mutated codons) represent mutational hotspots as they are the site of recurrent missense mutations (solid lines) that have been reported in three or more probands (mutational frequency of >1.5%), and/or affected by multiple (3 or more) different missense mutations (dashed lines). Mutations affecting these codons are clustered in three regions, which are the 2nd peptide loop of the extracellular domain, venus flytrap (VFT) cleft region Ca2+o binding site, and the region encompassing transmembrane domains (TMD) 6 and 7. Codons 173, 221, 297 and 802 (thickened lines) are the site of missense mutations that may lead to a loss- or gain-of-function and are termed ‘switch’ codons or residues. (B) Homology modelling of the CaSR VFT cleft region ligand-bound Ca2+o binding site based on the crystal structure of the metabotropic glutamate receptor type 1. The Leu173 (L173) and Pro221 (P221) switch residues are predicted to be located in short α-helices within lobes 1 and 2, respectively, that form the entrance to the Ca2+o binding site within the VFT cleft. The L173 and P221 side chains are shown in purple, the side chains of predicted Ca2+o binding residues (Ser147 (S147), Ser170 (S170), Asp190 (D190), Tyr218 (Y218) and Glu297 (E297)) are shown in cyan, and a bound calcium ion is shown as a green sphere. The side chains of L173 and P221 are predicted to extend across the entrance to the Ca2+o binding site. Mutations affecting these residues may lead to opposing effects on CaSR function by influencing the entry and binding of calcium within the VFT cleft region. Adapted, with permission, from Hannan FM, Nesbit MA, Zhang C, Cranston T, Curley AJ, Harding B, Fratter C, Rust N, Christie PT, Turner JJ, et al. (2012) Identification of 70 calcium-sensing receptor mutations in hyper- and hypo-calcaemic patients: evidence for clustering of extracellular domain mutations at calcium-binding sites. Human Molecular Genetics 21 2768–2778. Copyright 2012 Oxford University Press.
Familial disorders of Ca2+o sensing.
Familial hypocalciuric hypercalcaemia type 1 (FHH1)
FHH comprises three genetically distinct conditions, designated as FHH types 1–3 (Table 1), which are due to loss-of-function mutations affecting the CaSR, Gα11 and AP2σ proteins, respectively (Pollak et al. 1993, Nesbit et al. 2013a,b). FHH1 (OMIM #145980) is the most common type and accounts for ~65% of all FHH cases (Hannan et al. 2012). FHH is a highly penetrant autosomal dominant condition characterised by lifelong non-progressive mild-to-moderate hypercalcaemia, normal (in ~80% of patients) or mildly raised serum PTH levels (in ~20% of patients) (Firek et al. 1991), and low urinary calcium excretion (in ~95% of patients) (Marx 2015). FHH is considered to be a benign disorder, as patients are typically asymptomatic (Hannan & Thakker 2013). However, an increased prevalence of chondrocalcinosis with advancing age (Volpe et al. 2009) and occasional cases of pancreatitis have been reported (Pearce et al. 1996c). Individuals with FHH have been misdiagnosed as having PHPT and undergone parathyroidectomy, which generally fails to normalise the hypercalcaemia (Hannan & Thakker 2013). Mutational analysis may be required to distinguish FHH from PHPT, and to date, FHH1 has been associated with >130 different mutations of the CASR gene (Fig. 3), which are missense substitutions in >85% of cases, whereas nonsense, deletion, insertion and splice-site mutations that lead to truncated CaSR proteins have been described in <15% of cases (Hannan et al. 2012, Hannan & Thakker 2013). Studies of FHH1-associated CaSR mutations have identified critical receptor structure–function relationships and demonstrated a mutational hotspot within the ECD. Indeed, an analysis of the locations of recurrent FHH1-causing CaSR mutations or residues affected by multiple different loss-of-function CaSR mutations has revealed a clustering of mutations at the major predicted Ca2+o binding site located within the cleft region of the bilobed extracellular VFT domain of the CaSR (Fig. 3) (Hannan et al. 2012). These mutated residues may be directly involved in the binding of Ca2+o or indirectly influence alterations in receptor conformational states that occur upon Ca2+o binding (Huang et al. 2007, 2009, Hannan et al. 2012). Moreover, in vitro functional expression studies have identified specific VFT domain residues, which when mutated can result in opposing effects on CaSR responses and lead to a loss or gain of function (Fig. 3) (Hannan et al. 2012, Zhang et al. 2014). These residues may potentially act as intra-molecular switches to regulate the entry and binding of Ca2+o within the VFT cleft region (Fig. 3) (Hannan et al. 2012, Zhang et al. 2014). FHH-causing mutations also cluster in the CaSR transmembrane domain (TMD) and may inhibit the transmission of activation signals to the intracellular environment, by impairing interactions with heterotrimeric G proteins and other components of the CaSR signal transduction pathway (Leach et al. 2012). Some loss-of-function CaSR mutations have been shown to cause signalling bias by switching the wild-type CaSR from preferentially coupling with intracellular Ca2+ (Ca2+i) to a mutant receptor that signals equally via the Ca2+i and MAPK pathways, or which predominantly acts via MAPK (Leach et al. 2012). Loss-of-function CaSR mutations causing signalling bias are located within the ECD and TMD; however, the structural motifs within these regions that determine whether the CaSR preferentially couples to Ca2+i or MAPK pathways remain to be established. Around 50% of CaSR mutations that lead to FHH1 have been shown to result in reduced cell-surface receptor expression as a consequence of defective trafficking to the plasma membrane, with mutant CaSRs being retained intracellularly and unable to exit either the endoplasmic reticulum or Golgi apparatus (Huang & Breitwieser 2007, White et al. 2009). Such cellular studies also provide an explanation for the benign phenotype of FHH1, which is a heterozygous condition, by demonstrating that co-expression of wild-type and mutant FHH1-causing CaSRs ameliorates the loss of function, with the co-expressed wild-type CaSRs increasing trafficking of mutant receptors to the plasma membrane via the ADIS mechanism (Grant et al. 2012). Whereas, cells expressing only mutant loss-of-function CaSRs have both defective membrane targeting and reduced signalling responses (Grant et al. 2012).
Neonatal severe hyperparathyroidism (NSHPT)
NSHPT (OMIM #239200) is a potentially life-threatening disorder most often caused by homozygous or compound heterozygous loss-of-function CaSR mutations (Table 1) (Chattopadhyay & Brown 2006, Hannan et al. 2012, Hannan & Thakker 2013). NSHPT is characterised by severe neonatal hypercalcaemia (serum calcium concentrations are typically 3.5–5.0 mmol/L), 5- to 10-fold elevations of serum PTH concentrations, marked parathyroid gland enlargement, failure to thrive, and hyperparathyroid skeletal disease leading to multiple fractures and respiratory distress (Hannan & Thakker 2013, Murphy et al. 2016). Parathyroidectomy is usually required to treat NSHPT, and bisphosphonates have been successfully used in some patients to manage marked hypercalcaemia and skeletal demineralisation before parathyroid surgery (Waller et al. 2004, Wilhelm-Bals et al. 2012). Severe neonatal hypercalcaemia is also reported to be associated with heterozygous loss-of-function CaSR mutations, and these findings indicate that NSHPT may be due to factors other than mutant gene dosage. For example, the degree of severity of a dominant-negative mutation or maternal serum calcium concentration may play a role in the phenotypic expression of a CaSR mutation in the neonate (Hannan & Thakker 2013). More than 25 different CaSR mutations have been described in association with NSHPT, of which >40% are either nonsense or frameshift mutations that are predicted to lead to a truncated CaSR (Hannan et al. 2012, Hannan & Thakker 2013).
Primary hyperparathyroidism (PHPT) and marked hypercalcaemia presenting after infancy
Loss-of-function CaSR mutations may occasionally present after the neonatal period with marked hypercalcaemia. Indeed, homozygous loss-of function mutations, which are located at the N-terminal region of the CaSR, have been reported to lead to symptomatic hypercalcaemia in childhood or early adulthood, which required treatment with parathyroidectomy (Chikatsu et al. 1999, Miyashiro et al. 2004). Occasionally, heterozygous and homozygous loss-of function CaSR mutations have been detected in adult patients with PHPT caused by parathyroid adenomas or hyperplasia (Table 1) (Hannan et al. 2010a). The occurrence of PHPT or severe FHH after infancy may be due to the degree of loss of function associated with the underlying CaSR mutations. Indeed, the homozygous CaSR mutations, present in these patients, have been associated with milder alterations in Ca2+i signalling than homozygous mutations leading to NSHPT (Hannan et al. 2010a). An analysis of CaSR mutations identified in patients with PHPT or severe FHH that presented after infancy has indicated that CaSR mutations located in the receptor ECD are associated with more severe hypercalcaemia. This hypercalcaemia is recalcitrant to treatment by parathyroidectomy, possibly due to multiple gland involvement (Hannan et al. 2010a). These findings may have some clinical utility in identifying those patients (i.e. with CaSR ECD mutations) in whom a full neck exploration is required, as they are more likely to have multi-gland disease (Hannan et al. 2010a, Hannan & Thakker 2013).
Autosomal dominant hypocalcaemia type 1 (ADH1) and Bartter syndrome type V
ADH comprises two genetically distinct disorders, designated as ADH types 1 and 2 (Table 1), which are caused by germline gain-of-function mutations of the CaSR and Gα11 proteins, respectively (Pollak et al. 1994, Pearce et al. 1996b, Nesbit et al. 2013a). ADH1 (OMIM #601198) accounts for ~70% of all ADH cases (Nesbit et al. 2013a). ADH is characterised by mild-to-moderate hypocalcaemia, with serum calcium concentrations, adjusted for variations in serum albumin concentrations, rarely below 1.50 mmol/L, and ~50% of patients may develop hypocalcaemic symptoms such as paraesthesia, carpopedal spasms and seizures (Pearce et al. 1996b, Hannan & Thakker 2013, Nesbit et al. 2013a). Although ADH is associated with increased circulating phosphate concentrations and inappropriately low or normal PTH concentrations, this is considered to represent a distinct disease entity from hypoparathyroidism. This is because affected individuals generally have PTH concentrations that are detectable and within the reference range and also a relative hypercalciuria that is characterised by urinary calcium-to-creatinine ratios that are within or above the reference range (Pearce et al. 1996b, Hannan & Thakker 2013, Nesbit et al. 2013a). Ectopic calcification of the kidneys and basal ganglia is a common feature of ADH and affects >35% of patients (Pearce et al. 1996b, Hannan & Thakker 2013, Nesbit et al. 2013a). Patients with severe gain-of-function CaSR mutations may also have Bartter syndrome type 5, which is characterised by hypokalaemic alkalosis, renal salt wasting and hyperreninaemic hyperaldosteronism (Vargas-Poussou et al. 2002, Watanabe et al. 2002, Kinoshita et al. 2014). Mutational analysis is commonly required for the diagnosis of ADH, and >70 different CaSR mutations have been identified to date in individuals affected with ADH1 (Hannan et al. 2012, Hannan & Thakker 2013). Of these mutations, 95% are heterozygous missense substitutions (Hannan & Thakker 2013). The structure–function analyses of these mutations have defined key peptide motifs of the CaSR that maintain this receptor in an inactive conformation. In particular, gain-of-function mutations cluster within the second peptide loop of the extracellular VFT domain (residues 116–136) (Fig. 3) (Jensen et al. 2000) that contributes to the interface of the dimeric CaSR (Hu & Spiegel 2007, Zhang et al. 2016). Mutations affecting this extracellular peptide loop may lead to a gain of function by promoting conformational changes such as dimer rotation that facilitates receptor activation (Hu & Spiegel 2007). A second hotspot for ADH1-associated mutations is located in a region that encompasses transmembrane domains 6 and 7, and the intervening third extracellular loop of the CaSR (residues 819–837) (Fig. 3) (Hu et al. 2005). This region is likely important for locking the ligand-unbound family C GPCRs in an inactive conformation by forming a network of interactions with other transmembrane domains (Dore et al. 2014) that inhibits the binding of G proteins. Cellular studies have demonstrated that most ADH1-causing CaSR mutations cause a signalling bias by coupling more strongly to Ca2+i mobilisation than to the MAPK pathway, which contrasts with FHH1-causing CaSR mutations, which are biased towards MAPK signalling (Leach et al. 2012).
Use of calcimimetic and calcilytic drugs for disorders caused by CaSR mutations
Calcimimetics are ligands that mimic or enhance the effects of Ca2+o at the CaSR and are divided into two types: type I calcimimetics are agonists, which include naturally occurring ligands such as polyvalent cations; and type II calcimimetics are positive allosteric modulators that include l-amino acids and cinacalcet, which is a synthetic amino alcohol compound (Nemeth et al. 2004). Calcilytics are inhibitors of CaSR function, and to date, no endogenous calcilytic compounds have been identified. However, synthetic calcilytic compounds have been generated and shown to act as negative allosteric modulators of the CaSR (Nemeth & Shoback 2013). A recent study has identified a putative binding cavity for synthetic calcimimetic and calcilytic compounds within the extracellular portion of the CaSR TMD and delineated key glutamate residues within the binding cavity, which may facilitate the conformational changes that lead to altered receptor activity upon the binding of these compounds (Leach et al. 2016). Calcimimetic drugs represent a potential targeted therapy for NSHPT and symptomatic forms of FHH1. They are shown to improve the signalling by loss-of-function CaSR mutants in vitro (Rus et al. 2008, Leach et al. 2013) and to act as pharmacochaperones that facilitate correct protein folding and plasma membrane targeting of mutant CaSRs (Huang & Breitwieser 2007, White et al. 2009). These in vitro effects of calcimimetic drugs are in keeping with reports of FHH1 patients with marked hypercalcaemia or complications such as recurrent pancreatitis who have responded to treatment with cinacalcet, which is licensed for the management of certain hyperparathyroid disorders (Timmers et al. 2006, Festen-Spanjer et al. 2008). Cinacalcet has also been successfully used to manage life-threatening hypercalcaemia in NSHPT probands harbouring a heterozygous CaSR mutation, Arg185Gln (Gannon et al. 2014). However, it is ineffective for NSHPT caused by biallelic deletional CaSR mutations (Atay et al. 2014).
Symptomatic ADH is conventionally treated with calcium and active vitamin D preparations. However, these therapies do not rectify the underlying pathophysiological alterations in parathyroid and renal tubular function, and their use predisposes affected individuals to the development of marked hypercalciuria, nephrocalcinosis, nephrolithiasis and renal impairment (Pearce et al. 1996b, Hannan & Thakker 2013, Nesbit et al. 2013a). ADH1 patients have also been treated with recombinant PTH (1–34) (teriparatide); however, when administered by once- or twice-daily bolus injections, this peptide may not always prevent hypercalciuric renal complications (Theman et al. 2009). Calcilytic drugs, which are CaSR negative allosteric modulators, represent a potential targeted therapy for ADH1. Calcilytics comprise two main classes of compounds, which are the amino alcohols (e.g. NPS-2143, ronacaleret and JTT-305/MK-5442) and quinazolinones (e.g. ATF936 and AXT914) (Nemeth & Goodman 2016). In vitro studies have shown that NPS-2143, a long-acting calcilytic, corrects the gain of function associated with ADH-causing CaSR mutations (Hu et al. 2005, Letz et al. 2010, Lia-Baldini et al. 2013, Hannan et al. 2015b). However, the in vitro efficacy of NPS-2143 was reduced by mutations affecting NPS-2143-binding residues within the TMD (Hu et al. 2005, Letz et al. 2010). In contrast, the quinazolinone calcilytic drugs (ATF936 and AXT914) have been demonstrated to rectify the excessive signalling responses of all ADH mutants evaluated to date, including those mutations leading to constitutive activation and/or Bartter syndrome type 5 (Letz et al. 2014). To assess whether calcilytics may ameliorate the hypocalcaemia associated with ADH1, these drugs have been administered to mouse models harbouring germline gain-of-function CaSR mutations. In a single-dose in vivo study, NPS-2143 was administered to Nuf mice, which have hypocalcaemia, reduced plasma PTH concentrations and ectopic calcification in association with a germline gain of function Casr mutation, Leu723Gln (Hough et al. 2004, Hannan et al. 2015b). Intraperitoneal injection of NPS-2143 significantly increased plasma calcium and PTH concentrations in heterozygous- and homozygous-affected Nuf mice at 1 h after administration, with values returning to baseline after 4 h. The elevations in plasma calcium induced by NPS-2143 were not associated with any increase in urinary calcium excretion (Hannan et al. 2015b). Longer-term in vivo studies involving the JTT-305/MK-5442 calcilytic compound have been undertaken in two ADH1 mouse models, which harbour germline Cys129Ser and Ala843Glu gain-of-function CaSR mutations, respectively (Dong et al. 2015). Administration of JTT-305/MK-5442 by daily oral gavage over a 12-week period led to sustained increases in serum calcium concentrations and a significant reduction in urinary calcium excretion in both ADH1 mouse mutants (Dong et al. 2015). Recently, a calcilytic compound known as NPSP795 has been evaluated in a clinical trial involving five ADH1 patients. The intravenous administration of NPSP795 significantly increased plasma PTH concentrations and reduced urinary calcium excretion (Ramnitz et al. 2015). However, circulating calcium levels were not altered in this study. The optimal dosing regimen for NPSP795, which is a short-acting calcilytic compound intended to elicit a rapid and transient increase in plasma levels of PTH, remains to be established in ADH1 patients (Ramnitz et al. 2015).
Disorders associated with G-protein α-11 subunit mutations
Germline mutations affecting Gα11, which is encoded by the GNA11 gene (Fig. 4) on chromosome 19p13.3, have recently been identified as the genetic cause of FHH and ADH in some patients. This finding has revealed Gα11 to be a major component of the CaSR signalling pathway and highlighted its importance in Ca2+o homeostasis. Loss-of-function Gα11 mutations give rise to FHH2 (Nesbit et al. 2013a, Gorvin et al. 2016), whereas germline gain-of-function Gα11 mutations are associated with ADH2 (Fig. 4 and Table 1) (Mannstadt et al. 2013, Nesbit et al. 2013a, Li et al. 2014, Piret et al. 2016) and somatic gain-of-function Gα11 mutations cause uveal melanomas (Van Raamsdonk et al. 2010).
(A) Schematic representation of the genomic organisation of the GNA11 gene showing germline disease-associated mutations. The GNA11 gene consists of 7 coding exons (1–7), and the start (ATG) and stop (TGA) codon are in exons 1 and 7, respectively. The 5′ portion of exon 1 and the 3′ portion of exon 7 are untranslated (open boxes). The 3′ portion of exon 2, exon 3 and 5′ portion of exon 4 encode the Gα11 helical domain, which is connected by two short peptides, termed linker 1 (L1) and linker 2 (L2), to the GTPase domain. The Gα11 GTPase domain is encoded by the 3′ portion of exon 1, 5′ portion of exon 2, 3′ portion of exon 4 and exons 5–7. Three flexible regions, termed switch regions 1–3 (S1–S3, shown in blue), which undergo conformational changes during Gα11 activation are encoded by exons 4 and 5. The location of reported FHH2- and ADH2-causing mutations is shown. (B) Three-dimensional homology model of Gα11 showing the location of residues mutated in FHH2 (blue) and ADH2 (red). The homology model is based on the crystal structure of Gαq (PDB accession number 3AH8) (Nishimura et al. 2010), which shares 90% amino acid identity with Gα11. The Gα11 helical and GTPase domains are shown bound to GDP aluminium fluoride (GDP-AlF4, green), which is a non-hydrolysable analogue of GTP. The three flexible switch regions are highlighted in cyan, and the L1 and L2 peptides are shown in yellow. The β2–β3 hairpin loop, which comprises part of the Gα–GPCR interface, is shown in orange. Adapted, with permission, from Nesbit MA, Hannan FM, Howles SA, Babinsky VN, Head RA, Cranston T, Rust N, Hobbs MR, Heath H 3rd & Thakker RV (2013) Mutations affecting G-protein subunit alpha11 in hypercalcemia and hypocalcemia. New England Journal of Medicine 368 2476–2486. Copyright 2013 Massachusetts Medical Society.
Familial hypocalciuric hypercalcaemia type 2 (FHH2)
The existence of a genetically distinct form of FHH was first highlighted by genetic linkage studies that mapped an FHH locus to chromosome 19p, which contains the GNA11 locus. This form of FHH was designated as FHH2 (OMIM #145981) (Table 1) (Heath et al. 1993). GNA11 was considered a likely candidate gene for FHH2, as this encodes the Gα11 protein, which is highly expressed in the parathyroid glands (Varrault et al. 1995) and acts as a signalling partner for the CaSR (Hofer & Brown 2003). Moreover, mouse model studies have demonstrated parathyroid specific ablation of Gα11, and the related Gαq protein, to result in marked hypercalcaemia, hyperparathyroidism and parathyroid gland enlargement (Wettschureck et al. 2007). DNA sequence analysis of the reported FHH2 kindred revealed a germline heterozygous GNA11 mutation that resulted in an in-frame isoleucine deletion at codon 199 or 200 (Ile199/200del) of the Gα11 protein (Nesbit et al. 2013a). Mutational analysis of the GNA11 gene in additional FHH probands, who did not harbour CASR mutations, has identified heterozygous Leu135Gln and Thr54Met missense mutations in two unrelated probands (Nesbit et al. 2013a, Gorvin et al. 2016). The Thr54Met, Leu135Gln and Ile199/200del Gα11 mutations are associated with a mild FHH phenotype characterised by serum adjusted calcium concentrations <2.80 mmol/L (Nesbit et al. 2013a, Gorvin et al. 2016). In keeping with these clinical findings, in vitro studies have shown that these FHH2-associated Gα11 mutations lead to a mild impairment of CaSR signal transduction (Nesbit et al. 2013a, Gorvin et al. 2016). Indeed, the Thr54Met, Leu135Gln and Ile199/200del FHH2 mutants were associated with around a 30% increase in the half-maximal effective concentration (EC50) of CaSR-expressing cells, whereas CaSR mutations leading to FHH1 generally cause a >50% increase in the EC50 value (Bai et al. 1996, Pearce et al. 1996a, Hannan et al. 2012).
Homology modelling revealed that FHH2-causing mutations are located within key regions of the Gα11-subunit, which consists of a GTPase domain that binds GDP and GTP and a smaller helical domain that acts as a clasp to secure these bound guanine nucleotides (Fig. 4) (Oldham & Hamm 2008). Thus, the Ile199/200del mutation is located within the GTPase domain and predicted to disrupt a hairpin loop, which comprises part of the Gα-GPCR interface and is also situated between flexible ‘switch’ regions (Fig. 4) that undergo substantial conformational changes upon GTP binding (Nesbit et al. 2013a). In contrast, the Leu135Gln mutation is located in the Gα11 helical domain (Fig. 4) (Nesbit et al. 2013a). It is not predicted to influence CaSR–Gα11 coupling, but instead likely diminishes CaSR signal transduction by influencing the interaction of Gα11 with downstream effectors. The Thr54Met mutation is located at the interface between GTPase and helical domains (Fig. 4) and predicted to impair coupling and/or dissociation of Gα11 from the CaSR by influencing guanine nucleotide binding at the inter-domain interface (Gorvin et al. 2016). Thus, the identification of these FHH2-causing Gα11 mutations has revealed residues critical for Gα11-subunit function.
Autosomal dominant hypocalcaemia type 2 (ADH2)
After the identification of loss-of-function Gα11 mutations leading to FHH2, it was hypothesised that gain-of-function germline Gα11 mutations may have opposite effects on Ca2+o homeostasis and give rise to a disorder with an ADH-like phenotype. DNA sequence analysis of eight ADH probands, who did not harbour CaSR mutations, identified germline heterozygous Gα11 mutations in two individuals (Nesbit et al. 2013a). In vitro functional studies of these mutations, which comprised Arg181Gln and Phe341Leu Gα11 missense substitutions, demonstrated cells expressing the mutant Gα11 proteins to have enhanced sensitivity to Ca2+o, consistent with a gain of function (Nesbit et al. 2013a). In parallel with these studies, genetic linkage studies of two unrelated hypocalcaemic kindreds mapped the disease locus to chromosome 19p13.3, which is the location of the GNA11 locus. DNA sequence analysis revealed the occurrence of germline heterozygous Gα11 mutations, Arg60Cys and Ser211Trp (Mannstadt et al. 2013). Moreover, heterozygous Arg60Leu and Val340Met Gα11 mutations have been identified by whole-exome sequencing in additional ADH kindreds, and these mutations were demonstrated to lead to enhanced CaSR-mediated signal transduction (Li et al. 2014, Piret et al. 2016). These individuals and families with gain-of-function Gα11 mutations, who were designated as having ADH2 (OMIM #615361) (Table 1), generally had serum-adjusted calcium concentrations ranging 1.75–2.15 mmol/L. The affected individuals typically presented with hypocalcaemic symptoms such as paraesthesia, muscle cramps, carpopedal spasm and seizures (Mannstadt et al. 2013, Nesbit et al. 2013a, Li et al. 2014, Piret et al. 2016). Some ADH2 patients were susceptible to treatment-related hypercalciuria, nephrocalcinosis and nephrolithiasis (Li et al. 2014, Piret et al. 2016), although affected individuals generally had a milder urinary phenotype, with significantly reduced urinary calcium excretion compared with ADH1 patients who harbour gain-of-function CaSR mutations (Li et al. 2014). Furthermore, patients with germline gain-of-function Gα11 mutations, in contrast to patients with gain-of-function CaSR mutations, harbour non-calcitropic phenotypes. For example, studies of the kindred with the Arg60Leu Gα11 mutation showed this to be associated with impaired post-natal growth, and the affected adults were significantly shorter than unaffected adult family members (height >2SD below mean of unaffected individuals) (Li et al. 2014). In addition, some affected members of the kindred with the Val340Met Gα11 mutation were found to have keratoconus, a corneal disorder (Piret et al. 2016).
In contrast to germline gain-of-function Gα11 mutations, which affect Ca2+o homeostasis, somatic gain-of-function Gα11 mutations are reported to cause uveal melanoma, a primary intraocular tumour, by inducing constitutive upregulation of proliferative signalling involving extracellular signal-regulated kinases 1 and 2 (ERK1/2) (Van Raamsdonk et al. 2010), which are components of the MAPK signalling pathway. However, in vitro studies have shown that the ADH-causing Gα11 mutations do not have such oncogenic potential, and that these ADH-causing germline mutants phosphorylated ERK1/2 only in the presence of Ca2+o, and were thus not constitutively activating (Babinsky et al. 2016). Thus, the milder disturbance of signalling associated with the ADH2 mutants may provide an explanation for their occurrence as a post-natal phenotype that can be transmitted as an autosomal dominant disorder, in contrast to the uveal melanoma-associated constitutively activating G-protein mutation, Gln209Leu, which has been shown to be cytotoxic when expressed at high levels (Radhika & Dhanasekaran 2001) and is likely to be embryonically lethal if present within the germline.
An analysis of the location of ADH2-causing mutations revealed that mutated Arg60 and Arg181 residues are situated adjacent to the linker 1 and linker 2 peptides, respectively (Mannstadt et al. 2013, Nesbit et al. 2013a). The peptides are predicted to act as a hinge that connects the Gα11 GTPase domain with the helical domain, thereby allowing these two domains to form a clamshell around bound guanine nucleotides (Fig. 4). Thus, mutations affecting the Arg60 and Arg181 residues may lead to the opening of the Gα11 clamshell and induce G protein activation by promoting the exchange of GDP for GTP. The ADH2-causing Ser211Trp mutation is located within the region of Gα11, which binds to the Gβγ heterodimer, and this mutation may promote the dissociation of the Gα11 subunit, thus enhancing CaSR-mediated signal transduction (Mannstadt et al. 2013). The mutated Phe341 residue is located at the C-terminus of the Gα-subunit (Fig. 4) and forms a part of the cluster of phenylalanine residues, which likely stabilises GTP in a conformation required for its hydrolysis (Kaya et al. 2014, Sun et al. 2015), and the ADH2-causing Phe341Leu mutation is thus predicted to activate Gα11 by impairing the hydrolysis of GTP to GDP. In contrast to these predicted effects of the Phe341Leu mutation, the neighbouring Val340 residue is not involved in GDP/GTP exchange (Rasmussen et al. 2011, Sun et al. 2015), but instead may influence the stability of Gα-GPCR interactions (Piret et al. 2016).
Use of calcimimetic and calcilytic drugs for disorders caused by Gα11 mutations
Although calcimimetic and calcilytic compounds represented targeted therapies for patients with CaSR mutations resulting in symptomatic forms of FHH1 and ADH1, it was unclear if these CaSR allosteric modulators may rectify abnormalities of the downstream Gα11 protein, and thus, have potential benefit for FHH2 and ADH2 patients. Recent in vitro studies have revealed cinacalcet and NPS-2143 to correct the loss and gain of function associated with Gα11 mutations leading to FHH2 and ADH2, respectively (Babinsky et al. 2016). Indeed, siRNA knockdown studies showed that these CaSR allosteric modulators directly influence signalling mediated by the FHH2 and ADH2 mutant Gα11 proteins rather than by exerting indirect effects on endogenously expressed wild-type Gα11 proteins (Babinsky et al. 2016). However, some Gα11 mutations (Ile199/200del and Phe341Leu) showed diminished sensitivity to cinacalcet and NPS-2143 (Babinsky et al. 2016) and these differences in the sensitivities of the mutants to CaSR-targeted drugs may be explained by an analysis of the crystal structure of the G protein alpha-s (Gαs) complexed with the β2-adrenergic receptor (Rasmussen et al. 2011). The analysis showed that residues homologous to Ile199 and Phe341, in the related Gαs protein, are located within a hydrophobic pocket at the interface between GPCR and Gα-subunit (Fig. 5). Thus, Gα11 mutations located at the GPCR–Gα interface may potentially influence the efficacy of CaSR allosteric modulators (Babinsky et al. 2016). The NPS-2143 calcilytic compound was also shown to rectify the constitutive activation caused by a uveal melanoma-associated Gα11 mutation, and these findings suggest a potential therapeutic role for calcilytics in the management of this intraocular tumour (Babinsky et al. 2016).
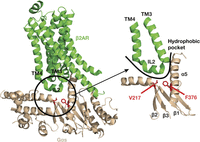
Three-dimensional model of Gαs (brown) bound to the β2-adrenergic receptor (β2AR, green) showing location of residues homologous to the Gα11 Ile199 and Phe341 residues, which are mutated in FHH2 and ADH2, respectively. Gαs residues homologous to the Gα11 Ile199 and Phe341 residues (red) are located within a hydrophobic region at the GPCR–Gα interface (black open circle). The β2AR–Gαs interface is formed by residues located within the β1 strand, hairpin loop linking the β2 and β3 strands, and the α5 helix of the Gαs protein, which interact with intracellular loop 2 (IL2) of the β2AR. The Gαs Val217 (V217) and Phe376 (F376) residues, which are homologous to the Gα11 Ile199 and Phe341 residues, comprise part of a hydrophobic pocket (curved line) at the Gα-subunit surface, which facilitates the docking of the β2AR IL2 with the Gαs protein (Rasmussen et al. 2011).
Disorders associated with adaptor-related protein complex 2 sigma subunit mutations
Familial hypocalciuric hypercalcaemia type 3 (FHH3)
Genetic linkage studies of two multi-generational FHH-unrelated kindreds from Oklahoma and Northern Ireland, designated FHHOK and FHHNI, respectively (McMurtry et al. 1992, Lloyd et al. 1999, Hannan et al. 2010b, Nesbit et al. 2010), mapped the disease locus, designated FHH3, to chromosomal 19q13.3, thereby highlighting a third genetically distinct form of FHH (FHH3, OMIM #600740) (Table 1). FHH3 was associated with elevations in plasma PTH, mild hypophosphataemia and osteomalacia in affected family members aged more than 30 years (McMurtry et al. 1992, Nesbit et al. 2010). Whole-exome capture and high-throughput sequence analysis revealed that affected individuals from the unrelated FHHOK and FHHNI kindreds harbour the same heterozygous germline Arg15Cys mutation of the adaptor-related protein complex 2 sigma subunit 1 (AP2S1) gene, which encodes the σ-subunit of the ubiquitously expressed heterotetrameric AP2 complex (Nesbit et al. 2013b). The AP2 complex is a central component of clathrin-coated vesicles and facilitates the endocytosis of plasma membrane proteins such as GPCRs (Kim & Benovic 2002). To date, AP2S1 mutations have been reported in >60 FHH3 patients and families. All affected individuals harbour a heterozygous missense mutation affecting the Arg15 residue of the encoded AP2σ-subunit (Fig. 6) and resulting in Arg15Cys, Arg15His or Arg15Leu (Fujisawa et al. 2013, Nesbit et al. 2013b, Hendy et al. 2014, Hannan et al. 2015a, Vargas-Poussou et al. 2016). Crystallography studies have revealed that the Arg15 residue plays a key role in binding to membrane cargo proteins (Fig. 6) (Kelly et al. 2008). It is predicted that these FHH3-causing Arg15 mutations disrupt an interaction between the AP2 complex and the intracellular carboxyl terminus of the CaSR, thereby impairing endocytosis of this GPCR (Nesbit et al. 2013b). This hypothesis is supported by in vitro expression studies, which have demonstrated that these FHH3-causing AP2σ mutations alter CaSR cell-surface expression and impair signal transduction in a dominant-negative manner (Nesbit et al. 2013b, Hannan et al. 2015a). Thus, these studies have revealed that the AP2 endocytic complex plays a role in the regulation of Ca2+o homeostasis.
(A) Schematic representation of the genomic organisation of the AP2S1 gene showing the location of FHH3-causing mutations. The AP2S1 gene consists of 5 coding exons (1–5), and the start (ATG) and stop (TGA) codon are in exons 1 and 5, respectively. The 5′ portion of exon 1 and the 3′ portion of exon 5 are untranslated (open boxes). The AP2σ protein is encoded by the 3′ portion of exon 1, exons 2, 3, 4, and the 5′ portion of exon 5 (dark grey). The FHH3-causing mutations all affect the Arg15 residue and comprise Arg15Cys (R15C), Arg15His (R15H) and Arg15Leu (R15L) missense substitutions. (B) Three-dimensional model of the heterotetrameric AP2 complex, which comprises α- (purple), β- (yellow), μ- (light blue) and σ- (light brown) subunits (PDB accession number 2JKR, (Kelly et al. 2008)). The AP2 complex is bound to a cargo protein recognition motif (green) via key polar contacts (shown in the red dashed circle) involving the AP2σ Arg15 (R15) residue (dark blue) and Arg21 (R21) residue of the AP2α-subunit. Adapted from Nesbit MA et al. Nat Genet. 2013 45:93–97. Adapted, with permission, from Nesbit MA, Hannan FM, Howles SA, Reed AA, Cranston T, Thakker CE, Gregory L, Rimmer AJ, Rust N, Graham U, et al. (2013) Mutations in AP2S1 cause familial hypocalciuric hypercalcemia type 3. Nature Genetics 45 93–97.
Nucleotide substitutions affecting codon 15 of the AP2S1 gene are predicted to result in the replacement of the wild-type Arg residue with a mutant Cys, Gly, His, Leu, Pro or Ser residue. All of these potential missense AP2σ substitutions have been demonstrated to diminish CaSR signal transduction in vitro (Hannan et al. 2015a). However, to date, only Arg15Cys, Arg15His and Arg15Leu AP2σ mutations have been observed in FHH3 patients (Fig. 6) (Fujisawa et al. 2013, Nesbit et al. 2013b, Hendy et al. 2014, Hannan et al. 2015a, Vargas-Poussou et al. 2016), and the likely cause of this mutation bias has been revealed by cellular studies. These studies have shown that the non-observed Arg15Gly, Arg15Pro and Arg15Ser AP2σ mutants impair cell growth in vitro (Hannan et al. 2015a). Thus, a possible explanation for the absence of the Arg15Gly, Arg15Pro and Arg15Ser AP2σ mutations in patients is that these deleterious mutations are embryonically lethal, whereas the FHH3-causing AP2σ mutations (Arg15Cys, Arg15His and Arg15Leu) are tolerated and compatible with embryonic and post-natal survival (Hannan et al. 2015a).
FHH3 may be associated with symptomatic hypercalcaemia and reduced bone mineral density, and with cognitive deficits and/or behavioural disturbances in children harbouring the Arg15Cys or Arg15Leu AP2σ mutations (Hannan et al. 2015a). FHH3 is associated with a more severe biochemical phenotype than FHH1, which is characterised by significantly higher serum calcium and magnesium concentrations and a significantly reduced fractional excretion of calcium (Hannan et al. 2015a, Vargas-Poussou et al. 2016). Furthermore, an analysis of adults and children with AP2σ mutations, by one study, has revealed a genotype–phenotype correlation with Arg15Leu probands having significantly higher serum calcium concentrations and presenting at a younger age, typically in childhood, compared with probands with Arg15Cys or Arg15His mutations (Hannan et al. 2015a). The absence of such a genotype–phenotype correlation in another study (Vargas-Poussou et al. 2016), which consisted of mainly adult FHH3 patients, suggests that the more severe hypercalcaemia associated with the Arg15Leu mutation might be age dependent.
Use of cinacalcet for symptomatic hypercalcaemia caused by AP2σ mutations
Cinacalcet has been evaluated as a therapy for symptomatic hypercalcaemia associated with FHH3. In vitro studies revealed that this calcimimetic drug rectifies the significantly impaired intracellular calcium and MAPK responses associated with all three FHH3-causing AP2σ mutations (Howles et al. 2016). The administration of cinacalcet at a dose of 30–60 mg daily to three symptomatic FHH3 probands, each harbouring an Arg15Cys, Arg15His or Arg15Leu AP2σ mutation, led to >20% reductions in serum calcium concentrations and improved symptoms in all three FHH probands (Howles et al. 2016). These studies show that cinacalcet-mediated allosteric modulation of the CaSR can rectify the loss-of-function and symptomatic hypercalcaemia that are associated with the three types of FHH3-causing Arg15 AP2σ mutations. Cinacalcet has also been shown to correct the hypercalcaemia in a patient with chromosome 22q11.2 deletion syndrome who also had an Arg15Leu AP2σ mutation (Tenhola et al. 2015).
Autosomal dominant hypocalcaemia type 3: the search for AP2σ mutations
Germline coding-region mutations of the CASR and GNA11 genes, which enhance CaSR-mediated signal transduction, have been identified in around 70% of ADH cases (Hannan et al. 2012, Nesbit et al. 2013a). This raises the possibility that ADH patients who do not harbour such mutations may instead have abnormalities of the untranslated or non-coding regulatory regions of CASR and GNA11 or have mutations involving other mediators of Ca2+o homeostasis. It had been postulated that some patients with ADH might harbour AP2σ mutations, which enhance the sensitivity of CaSR-expressing cells to Ca2+o, and this putative form of ADH was designated ADH type 3 (ADH3) (Rogers et al. 2014). However, an analysis of the AP2S1 gene in 19 patients and families who were considered to have ADH but did not have mutations of CASR and GNA11, or of other genes associated with isolated hypoparathyroidism, such as PTH or GCMB, failed to identify coding-region mutations or copy number variants (CNVs) (Rogers et al. 2014). Moreover, investigations of 10 familial cases and 50 sporadic cases of isolated hypoparathyroidism for coding-region mutations or CNVs affecting AP2S1 did not identify any abnormalities (Lambert et al. 2014). Thus, these studies indicated that AP2σ mutations are unlikely to cause hypocalcaemic disorders such as ADH.
Conclusion
The identification and characterisation of gene abnormalities underlying FHH and ADH have led to the delineation of a parathyroid and renal G protein-coupled Ca2+o-sensing mechanism, which involves the CaSR, Gα11 and AP2σ proteins. The CaSR is shown to play a pivotal role in the regulation of Ca2+o concentrations, whereas the Gα11 protein appears to be a key mediator of downstream CaSR signal transduction, and the AP2σ protein is likely required for both CaSR signalling and trafficking. These studies have also provided new insights into the clinical phenotypes of Ca2+o-sensing disorders and revealed FHH3 to represent a distinct disorder of Ca2+o homeostasis, which is characterised by symptomatic hypercalcaemia, low BMD and cognitive deficits. Advances have been made in the treatment of FHH and ADH, and the calcimimetic and calcilytic drugs have been shown to be of potential benefit for managing symptomatic forms of these Ca2+o-sensing disorders.
Footnote
This review is based on the 2015 Dale Medal Lecture, presented by Prof. Rajesh Thakker at the Society for Endocrinology BES 2015, Edinburgh, UK.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of this review.
Funding
The work in the Academic Endocrine Unit is supported by the United Kingdom Medical Research Council (MRC) programme grants – G9825289 and G1000467, and National Institute for Health Research (NIHR) Oxford Biomedical Research Centre Programme. R V Thakker is a Wellcome Trust Investigator and NIHR Senior Investigator.
- Received 21 July 2016
- Accepted 8 August 2016
- Made available online as an Accepted Preprint 19 September 2016
- © 2016 Society for Endocrinology
 This work is licensed under a Creative Commons Attribution 3.0 Unported License.
This work is licensed under a Creative Commons Attribution 3.0 Unported License.










