
Thyroid hormone metabolism in innate immune cells
- Correspondence should be addressed to A Boelen; Email: a.boelen{at}amc.uva.nl
-
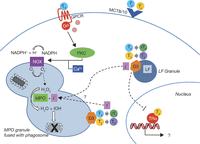 Figure 1
Figure 1Hypothetical pathways explaining the effects of thyroid hormone on neutrophil NAPDH oxidase activity and bacterial killing. Thyroid hormone induces neutrophil NADPH oxidase (NOX) activity, resulting in increased production of reactive oxygen species. This phenomenon is thought to be mediated via a non-genomic pathway involving binding of TH to a G-protein-coupled receptor (GPCR), which induces NAPDH oxidase activity. This effect is dependent on protein kinase C (PKC) and adequate intracellular Ca2+ levels (Mezosi et al. 2005). Intracellular thyroid hormone metabolism may also play a role in neutrophils during bacterial killing. The thyroid hormone-inactivating type 3 deiodinase (D3) is present in murine and human neutrophils (Boelen et al. 2005, 2008, van der Spek et al. 2016). Mice that lack this enzyme suffer from impaired bacterial killing (Boelen et al. 2009). D3 is located in the cytoplasm and in granules containing either myeloperoxidase (MPO) or lactoferrin (LF) (van der Spek et al. 2016). TH enters the neutrophil via transporters (MCT8 or MCT10) where it is inactivated by D3, which removes an iodine atom from the inner ring of the hormone, converting T4 to reverse (r)T3 and T3 to T2. Increased D3 activity therefore results in decreased intracellular levels of T3 together with the production of free iodide (I−). One hypothesis explaining the role of D3 in microbial killing is that the iodide produced by D3 is utilized by MPO together with hydrogen peroxidase (H2O2) to generate hypoiodite (IOH), a toxic compound that is capable of killing bacteria (Klebanoff 1967, Boelen et al. 2011). The reduction of intracellular T3 levels could theoretically also result in altered gene transcription, but no TH-responsive genes have been found in neutrophils yet.
-
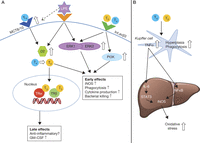 Figure 2
Figure 2Thyroid hormone induces a pro-inflammatory response in macrophages. (A) The effects of TH in macrophages are mediated through integrin αvβ3 or through modulation of intracellular TH levels. TH can bind to integrin αvβ3 on the macrophage cell surface resulting in the activation of PI3K and ERK1/2 pathways followed by the upregulation of inducible nitric oxide synthase (iNOS) (Chen et al. 2012). Alternatively, TH can enter the cell through TH transporters MCT8 or MCT10 after which the prohormone T4 is converted to active hormone T3 by type 2 deiodinase (D2) resulting in increased phagocytosis and cytokine response. D2 is induced in lipopolysaccharide (LPS)-stimulated macrophages (Kwakkel et al. 2014). The effects of intracellular T3 are partly mediated via TRα, which is required for adequate macrophage function (Billon et al. 2014, Kwakkel et al. 2014). Low-grade inflammation found in unstimulated TRα-knockout macrophages suggests that TRα possibly attenuates the rapid pro-inflammatory response generated by increased intracellular TH levels (Billon et al. 2014). (B) Kupffer cells are the resident macrophages of the liver. TH stimulation in vivo results in Kupffer cell hyperplasia and enhanced phagocytosis (Tapia et al. 1997, Valencia et al. 2004). TH transporter and receptor expression in Kupffer cells have not yet been studied. TH also increases the production of TNFα by Kupffer cells (Valencia et al. 2004, Fernandez et al. 2005, 2007b). TNFα produced by Kupffer cells results in liver NFκB activation (Valencia et al. 2004). IL-6 production is also increased, resulting in increased STAT3 activation (Tapia et al. 2006). The activation of both these pathways results in increased iNOS activity in the liver, resulting in the production of larger amounts of reactive oxygen species and hepatic oxidative stress (Fernandez et al. 2005).
-
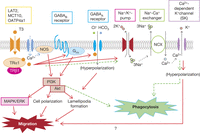 Figure 3
Figure 3Effects of thyroid hormone on microglia (reproduced with permission from Mori et al. 2015). Schematic representation of possible signal transduction pathways activated by T3. Microglia contain various TH transporters (LAT2, OATP4a1 and MCT10) and TH receptors. Intracellular T3 activates intracellular TRs (TRα1 and possibly TRβ1) and appears to also couple to various other factors, such as nitric oxide synthase (NOS), Gi/o-protein, PI3K and MAP/ERK. The receptors for gamma-aminobutyric acid (GABA) A and B are also involved in T3-induced microglial migration but not in T3-induced phagocytosis, the mechanism of which is unclear. The reverse mode of the Na+/Ca2+ exchanger (NCX) may be activated by Na+ influx by the Na+/K+ pump, resulting in Ca2+ influx, which in turn activates small-conductance Ca2+-dependent K+ channels (SK). This subsequently induces microglial migration and phagocytosis, possibly due to membrane hyperpolarization. Hypothetical signalling pathways for phagocytosis are indicated by the dotted line. Reproduced, with permission, from Mori Y, Tomonaga D, Kalashnikova A, Furuya F, Akimoto N, Ifuku M, Okuno Y, Beppu K, Fujita K, Katafuchi T, et al. (2015) Effects of 3,3’,5-triiodothyronine on microglial functions, Glia, volume 63, pages 906–920. Copyright (2015) Wiley Periodicals, Inc.
-
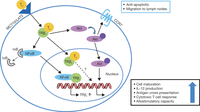 Figure 4
Figure 4Thyroid hormone enhances dendritic cell maturation and function. In dendritic cells, T3 enters the cell via TH transporters MCT10 or LAT2 and binds to cytoplasmic TRβ1 (Gigena et al. 2015 abstract 475, presented at the International Thyroid Congress). Upon binding of T3, TRβ1 translocates from the cytoplasm to the nucleus (Mascanfroni et al. 2008, 2010). T3 binding to TRβ1 also activates cytoplasmic NFκB by initiating degradation of IκB so that NFκB can translocate to the nucleus and regulate gene transcription, including induction of TRβ1 transcription, which is an NFκB target gene, thus forming a regulatory feedback loop controlling TRβ1 levels (Mascanfroni et al. 2008). Furthermore, binding of T3 to cytoplasmic TRβ1 activates the Akt pathway, leading to a nuclear shift of phosphorylated Akt and increased chemokine (C–C motif) receptor type 7 (CCR7) expression, which prolongs cell viability and augments cell migration towards lymph nodes (Mascanfroni et al. 2010, Alamino et al. 2015). T3 stimulation of DCs shifts the cells towards a more pro-inflammatory phenotype through induction of cell maturation, increased IL-12 production, improved antigen cross presentation and an enhanced ability to stimulate a cytotoxic T-cell response and trigger antigen-specific responses in vivo (Mascanfroni et al. 2008, 2010, Alamino et al. 2015, 2016).
- © 2017 Society for Endocrinology










